
ТЕСТ
1
-
Лекарственные
средства
– это вещества или их комбинации,
вступающие в контакт с организмом
человека или животного, проникающие в
органы, ткани организма человека или
животного, применяемые для профилактики,
диагностики (за исключением веществ
или их комбинаций, не контактирующих
с организмом человека или животного),
лечения заболевания, реабилитации, для
сохранения, предотвращения или прерывания
беременности и полученные из крови,
плазмы крови, из органов, тканей организма
человека или животного, растений,
минералов методами синтеза или с
применением биологических технологий. -
Фармацевтическая
субстанция –
лекарственное средство в виде одного
или нескольких обладающих фармакологической
активностью действующих веществ вне
зависимости от природы происхождения,
которое предназначено для производства,
изготовления лекарственных препаратов
и определяет их эффективность. -
Вспомогательные
вещества –
вещества неорганического или органического
происхождения, используемые в процессе
производства, изготовления лекарственных
препаратов для придания им необходимых
физико-химических свойств. -
Лекарственные
препараты –
лекарственные средства в виде
лекарственных форм, применяемые для
профилактики, диагностики, лечения
заболевания, реабилитации, для сохранения,
предотвращения или прерывания
беременности. -
Лекарственная
форма –
состояние лекарственного препарата,
соответствующее способам его введения
и применения и обеспечивающее достижение
необходимого лечебного эффекта. -
Дозировка
–
содержание одного или нескольких
действующих веществ в количественном
выражении на единицу дозы, или единицу
объема, или единицу массы в соответствии
с лекарственной формой либо для некоторых
видов лекарственных форм количество
высвобождаемого из лекарственной формы
действующего вещества за единицу
времени. -
Перечень
жизненно необходимых и важнейших
лекарственных препаратов –
утверждаемый Правительством Российской
Федерации перечень лекарственных
препаратов для медицинского применения,
обеспечивающих приоритетные потребности
здравоохранения в целях профилактики
и лечения заболеваний, в том числе
преобладающих в структуре заболеваемости
в Российской Федерации. -
Орфанные
лекарственные препараты – лекарственные
препараты, предназначенные исключительно
для диагностики или патогенетического
лечения редких заболеваний. -
Биологические
лекарственные препараты – лекарственные
препараты, действующее вещество которых
произведено или выделено из биологического
источника и для определения свойств и
качества которых необходима комбинация
биологических и физико-химических
методов. К биологическим лекарственным
препаратам относятся иммунобиологические
лекарственные препараты, лекарственные
препараты, полученные из крови, плазмы
крови человека и животного (за исключением
цельной крови), биотехнологические
лекарственные препараты, генотерапевтические
лекарственные препараты. -
Иммунобиологические
лекарственные препараты –
лекарственные препараты, предназначенные
для формирования активного или пассивного
иммунитета либо диагностики наличия
иммунитета или диагностики специфического
приобретенного изменения иммунологического
ответа на аллергизируюшие вещества. К
иммунобиологическим лекарственным
препаратам относятся вакцины, анатоксины,
токсины, сыворотки, иммуноглобулины и
аллергены. -
Биотехнологические
лекарственные препараты –
лекарственные препараты, производство
которых осуществляется с использованием
биотехнологических процессов и методов
(в том числе ДНК-рекомбинантной
технологии, технологии контролируемой
экспрессии генов, кодирующих биологически
активные белки в прокариотах и эукариотах,
включая измененные клетки млекопитающих),
гибридомного метода и метода моноклональных
антител. -
Генотерапевтические
лекарственные препараты –
лекарственные препараты, фармацевтическая
субстанция которых является рекомбинантной
нуклеиновой кислотой или включает в
себя рекомбинантную нуклеиновую
кислоту, позволяющую осуществлять
регулирование, репарацию, замену,
добавление или удаление генетической
последовательности. -
Наркотические
лекарственные средства –
лекарственные препараты и фармацевтические
субстанции, содержащие наркотические
средства и включенные в Перечень
наркотических средств, психотропных
веществ и их прекурсоров, подлежащих
контролю в Российской Федерации, в
соответствии с законодательством
Российской Федерации, международными
договорами Российской Федерации, в том
числе Единой конвенцией о наркотических
средствах 1961 года. -
Психотропные
лекарственные средства
– лекарственные препараты и
фармацевтические субстанции. Содержащие
психотропные вещества и включенные в
Перечень наркотических средств,
психотропных веществ и их прекурсоров,
подлежащих контролю в Российской
Федерации, в соответствии с законодательством
Российской Федерации, международными
договорами Российской Федерации, в том
числе Конвенцией о психотропных
веществах 1971 года. -
Радиофармацевтические
лекарственные средства –
лекарственные средства, которые содержат
в готовой для использования форме один
радионуклид или несколько радионуклидов.
ТЕСТ
2
-
При
валидации аналитических методик
количественного определения посторонних
примесей необходимо контролировать
специфичность,
предел количественного определения,
аналитическая область, линейность,
правильность, прецизионность.
-
Предел
обнаружения определяют
ПО=3,3*S/b
-
Аналитическая
область методики определения родственных
примесей находится
от
предела определения/обнаружения до
120%
-
Оборудование
на производстве работает должным
образом если пройдена
квалификация
-
Валидацию
необходимо проводить
при
введении нового промышленного регламента
-
Валидация
твердых лекарственных средств включает
в себя
распределение
частиц по размерам, насыпная плотность,
плотность при уплотнении
-
Какие
основные документы по валидации
протокол,
отчет
-
Масштабирование
с уменьшением
уменьшение
объема серии в ответ на падение рыночного
спроса
-
Изменение
каких параметров необходимо отслеживать
при масштабировании
состав
исходных материалов, процессы,
оборудование, производственная площадка,
размер производственной серии
-
Согласно
Биофармацевтической классификационной
системе к категории А относятся
активные
вещества с высокой проницаемостью и
хорошей растворимостью
-
Особенности
категории В
оценка
профиля растворения
-
Пострегистрационные
изменения уровня 2 (FDA)
изменения
могут оказать существенное влияние на
качество и эффективность лекарственной
формы
ТЕСТ
3
-
Уполномоченное
лицо
работник производителя лекарственных
средств -
Образование
Уполномоченного лица
высшее химическое -
Стаж
работы уполномоченного лица более
пяти лет -
Обязанности
уполномоченного лица подтверждение
того, что каждая импортируемая серия,
произведенная в РФ, соответствует
законодательству и требованиям
регистрационного досье -
Уполномоченное
лицо должно подтвердить, что процесс
производства осуществлен в соответствии
с приказом № 916 -
Кому
могут быть переданы обязанности
уполномоченного лица другому
уполномоченному лицу -
Концепция
уполномоченного лица финансируется
производителем -
Уполномоченное
лицо верифицирует
систему качества на предприятии -
Уполномоченное
лицо проводит подготовку
кадров предприятия -
Цель
концепции уполномоченного лица защита
потребителей лекарственных средств -
Ответственность
за выпуск некачественной продукции в
РФ лежит на уполномоченном
лице -
При
выпуске серии уполномоченное лицо
проводит ее сертификацию -
На
производстве уполномоченное лицо
делегирует
часть своих обязанностей ответственным
сотрудникам -
В
работе уполномоченного лица важно
вовлеченность
в производственный процесс -
Уполномоченное
лицо в административной структуре
предприятия имеет место в директорате -
Несколько
уполномоченных лиц на одном предприятии
может быть если на
предприятии есть в наличии несколько
производственных площадок -
Как
часто проводится повышении квалификации
уполномоченного лица не
реже 1 раза в 5 года -
«Соглашение
по качеству» регулирует определение
ответственности между
двумя уполномоченными лицами
ТЕСТ
4
-
Самоинспектирование
это репетиция
официального обследования -
Аудиты
систем качества по ISO
9000 не
заменяют сертификацию по GMP -
Внешний
аудит проводится для проверки
документов -
Аудит
третьей стороны проверка,
государством или негосударственным
сертификационным органом -
Жизненный
цикл управления по Шухарту-Демингу-Джурану
представляет собой цикл
«планируй – делай – проверяй – действуй» -
Внешний
аудит с привлечением сторонних
специалистов необходим для
малых предприятий с численностью не
более 25 человек -
Проверка
только инъекционных препаратов
называется инспектирование
в разрезе видов продукции -
Аудит
«Обзор документации» включает в себя
проверку электронные
системы ведения документации -
Проект
программы аудита согласовывается с
уполномоченным
лицом и руководителем службы качества -
План
выполнения аудита строится по следующей
схеме внесение
аудита в текущий план – выполнение
аудита – представление протокола –
исправление выявленных недостатков -
Как
расшифровывается термин «САРА» (КАПА)
программа
корректирующих и предупреждающих
действий -
Аудит
считается закрытым после выполнения
всех действий, предусмотренных планом
по устранению нарушений -
Можно
ли использовать прямые, наводящие или
утверждающие вопросы во время аудита?
Нет -
Итоговый
отчет о самоинспекции представляет
собой изложение
фактов, в основу которых положены
несоответствия, подкрепленные
объективными доказательствами -
Основополагающий
принцип аудита представляет собой если
что-то запланировано, то оно обязательно
и должно быть выполнено -
Аудитор
в процессе самоинспекции является
связующим звеном между руководством,
системой менеджмента качества и
персоналом -
Какие
требования к аудитору имеют большой
вес специальное
обучение по аудитам или большой опыт
проведения аудитов -
При
проведении аудита в течение двух дней
отчет необходимо заполнять ежедневно,
после окончания рабочего дня
ТЕСТ
5
1.
Менеджмент рисков это
действия по руководству и управлению
организацией в отношении риска
2.
В
течение какого времени необходимо
проводить менеджмент рисков:
в течении жизненного цикла фармацевтического
продукта
3.
Аббревиатура
НАССР расшифровывается как:
анализ опасностей и критических
контрольных точек
4.
НАССР пришел в фармацевтическую
промышленность из:
пищевой промышленности
5.
Риск это:
сочетание вероятности возникновения
опасности и серьезности его последствий
6.
Побочные эффекты лекарств это:
приемлемые опасности
7.
Критическая контрольная точка (ККТ)
это:
точка, где необходимо провести контроль
для предупреждения или ликвидации
опасности, или уменьшить ее до допустимого
уровня
8.
Критический предел (КТ) это:
критерий, который разделяет приемлемость
от неприемлемости
9.
Одним из основным инструментом менеджмента
риска является:
анализ дерева неисправностей (РТА)
10.
Сколько этапов в системе НАССР:
7
11.
Первым этапом в системе НАССР является:
проведение анализа опасностей
12.
Диаграмма Ишикавы это: причинно-следственная
диаграмма или «рыбий скелет»
13.
Инструмент «Дерево принятия решений»
применяется на этапе:
определения критических контрольных
точек
14.
Мониторинг для каждой критической
контрольной точки должен быть:
возможны оба варианта
15.
Когда необходимо применение коррективных
действий:
случае возникновения отклонения, с
целью возвращения ККТ под контроль
ТЕСТ
6
1.
Надлежащая лабораторная практика это:
GLP
2.
Какие процессы реализуются в ОКК:
контроль при рекламациях
3.
Стандартные операционные процедуры
(СОП)
имеют
форму:
письменных инструкций, разрабатываемых
и утверждаемых на данном предприятии
4.
Помещения должны быть максимально
удалены от источников вибрации,
электромагнитного излучения это
требования к помещениям, где расположены:
весы
5.
В аналитической лаборатории архивные
образцы хранятся:
в отдельном помещении
6.
Микробиологическая лаборатория и
лаборатория физико-химических методов
анализа
могут
быть расположены в одном здании если:
они находятся в разных помещениях
7.
Измерительное оборудование подлежит
поверке организациями, аттестованными:
Госстандартом
8.
Образец сравнения, полученный на
основании данных анализа стандартного
образца, используется:
для выполнения текущих анализов
9.
В лабораторных журналах на реактивы
должно быть указано:
происхождение реактива, условия хранения
и отпуска
10.
Проверка проб, стадий, операций – это:
Операционный контроль
11.
Материалы первичной упаковки:
непосредственно соприкасаются с
лекарственным препаратом
Сертификат GMP — это соблюдение изготовителем лекарственных препаратов требований надлежащей производственной практики. В России они сформулированы в национальном стандарте ГОСТ Р 52249-2009, который идентичен правилам, действующим в Европейском Союзе.
- К каким производствам применима эта процедура?
- Стандарт GMP в международной практике
- Правила GMP в России
- Процедура получения сертификата в России
- Стоимость получения сертификата
К каким производствам применима эта процедура?
В настоящее время в странах, которые контролируют соответствие стандарту GMP на своих территориях, его правила применяются для проверки качества следующих категорий продукции:
- лекарственные препараты;
- медицинские изделия различного назначения, включая те из них, которые применяются в диагностических целях;
- продукты питания и ингредиенты для их производства;
- биологически активные добавки.
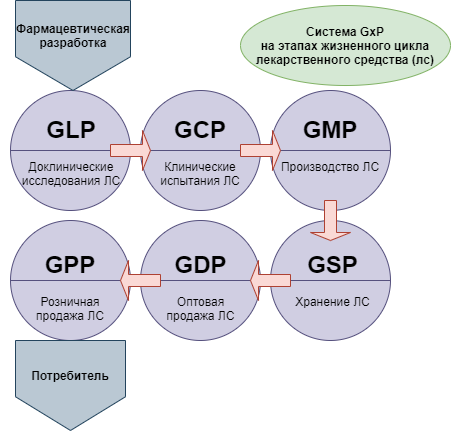
При этом для понимания ситуации следует принимать во внимание, что новая версия сертификации GMP — это не единственная система требований, которые в международной практике применяются в целях стандартизации медицинского обслуживания населения. Кроме них, производителям, работающим в такой сфере как фармация, необходимо соответствовать требованиям комплекса правил, объединенных под общим наименованием GxP:
- GLP — Good Laboratory Practice (надлежащая лабораторная практика);
- GCP — Good Clinical Practice (надлежащая клиническая практика);
- GDP — Good Distributon Practice (надлежащая дистрибьюторская практика);
- GACP — Good Agricultural and Collection Practice (надлежащая практика культивирования и сбора лекарственных растений).
Нормативная база
В Российской Федерации получение сертификата GMP осуществляется на основании действующей нормативной базы, включающей следующие основные правовые акты:
- национальный стандарт РФ ГОСТ Р 52249-2009, устанавливающий правила изготовления и контроля качества лекарственных препаратов;
- постановление Правительства от 5 июня 2008 года N 438 с рядом изменений, внесенных за последние годы, которое утверждает полномочия Министерства промышленности и торговли в этой области;
- постановление Правительства от 3 декабря 2015 года N 1314, устанавливающее порядок оценки соответствия производителей требованиям стандарта надлежащей практики;
- приказ Минпромторга от 14 июня 2013 года N 916, утверждающий правила применения надлежащей производственной практики в соответствии с актуальным стандартом;
- приказ Минпромторга от 26 мая 2016 года N 1714, определяющий административный регламент предоставления государственной услуги по выдаче документации, подтверждающей соответствие изготовителя установленным нормам надлежащей производственной практики;
- приказ Минпромторга России от 17.12.2015 N 4119, утверждающий правила ведения реестра сведений о том, какие лекарства имеют сертификат качества GMP в России.
При этом необходимо принимать во внимание, что в настоящий момент наша страна вместе с другими государствами, входящими в состав Евразийского экономического союза, находится на этапе становления общего рынка, объединяющего фармацевтическое и косметическое производство в границах Союза. Это предполагает в том числе введение в действие единых требований к качеству и безопасности таких продуктов. В соответствии с принятым в мире порядком они реализуются в форме внедрения стандартов надлежащей производственной практики. Применение таких стандартов регулируется следующими нормативными документами:
- Решение Совета ЕЭК от 3 ноября 2016 года N 77, утверждающее правила надлежащей производственной практики в границах ЕАЭС;
- Приказ Минпромторга от 4 сентября 2020 года N 2945, которым введен административный регламент предоставления госуслуги по выдаче документации, подтверждающей соответствие производств установленным правилам.
Обратите внимание!
Для полноценного применения разработанного административного регламента необходимо решение Правительства о порядке реализации некоторых процедур, связанных с проведением фармацевтических инспекций. Приказ № 2945 вступит в силу только после принятия соответствующего постановления: пока этого не произошло.
Преимущества обладания сертификатом
Несмотря на необходимость проведения достаточно сложной и дорогостоящей процедуры, производители знают, что сертификация по стандартам GMP является весьма важной для представителей фармацевтической отрасли. В частности, оно обеспечивает продукции и производству следующие серьезные преимущества:
- стабильное качество продукции, не зависящее от внешних факторов;
- повышение доверия потребителей, включая крупных оптовых покупателей, которые всегда отслеживают, какие производители имеют сертификат соответствия GMP на их продукцию;
- возможность вывода продукции на международные рынки, где ее может купить гораздо больше потребителей;
- возможность привлечения инвесторов для реализации проектов по расширению производства;
- получение преимуществ при участии в конкурсном отборе поставщиков, в том числе для государственных закупок.
КОММЕНТАРИЙ ЭКСПЕРТА АТТЭК
Каков срок действия сертификата?
Срок действия российских сертификатов составляет 3 года. При этом срок действия иностранного сертификата GMP составляет от 1 до 3 лет. По истечении этого периода сертификацию потребуется проходить заново. Кроме того, это означает, что на протяжении всего этого срока компании необходимо обеспечить соответствие своего производства и продукции требованиям комплекса правил GMP.
Кто в России занимается сертификацией по стандартам GMP?
Сейчас сертификация контролируется департаментом развития медицинской и фармацевтической промышленности Министерства промышленности и торговли РФ. Он является организацией, ответственной за обеспечение надлежащего контроля за качеством, безопасностью и эффективностью лекарственных средств. Осуществлением требуемых сертификационных процедур занимается Государственный институт лекарственных средств и надлежащих практик (ФГБУ «ГИЛС и НП»).
Стандарт GMP в международной практике
Процесс сертификации на соответствие лекарственного препарата стандартам GMP в международной практике имеет комплексный характер, а ее основной целью является подтверждение безопасности и действенности продукции. В этой связи для достижения поставленной цели специалисты аккредитованных сертификационных организаций не ограничиваются оценкой ряда выборочных образцов лекарственных препаратов, как это часто предусматривается другими стандартами. В процедуру установления требуемого уровня качества лекарств любой международный центр сертификации лекарственных средств включает оценку предприятия, занимающегося его выпуском. В результате эксперты, занимающиеся проведением сертификации, анализируют конкретный препарат и процесс его выпуска в следующих областях:
- оценка производства на соответствие критериям безопасности, включая проведение его проверки в отношении вероятности попадания в продукт посторонних примесей и веществ;
- оценка производства на соблюдение технических требований к выпуску продукции, включая выполнение условий относительно влажности, температуры и других параметров в производственных помещениях;
- оценка качества, безопасности и действенности лекарственных средств, производимых на конкретном предприятии;
- оценка соответствия параметров производства и характеристик лекарственного средства нормативной документации, принятой в рамках процедуры GMP.
Правила GMP в России
Порядок и сроки проведения всех операций в рамках этой процедуры, список лиц и организаций, ответственных за их осуществление, размер платы за проведение экспертной оценки и другие аспекты выполнения сертификации определены постановлением Правительства № 1314.
Процедура получения сертификата в России
Первым шагом для производителя, который желает пройти сертификацию, является подача соответствующего заявления в Минпромторг. В течение 10 рабочих дней специалисты ведомства проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
В случае необходимости они вправе запросить у заявителя дополнительные документы, которые он обязан предоставить в течение 20 рабочих дней. В случае, если в отношении данного препарата принято положительное решение о проведении процедуры сертификации, необходимые данные направляются в ФГБУ «ГИЛС и НП», который в течение 20 рабочих дней с момента их получения обязан определить дату проведения сертификационных мероприятий и внести ее в график. Такая дата должна наступить не позднее 160 рабочих дней со дня, когда специалисты Минпромторга приняли положительное решение о сертификации, а сама экспертиза и расшифровка ее результатов должны занимать не более 10 рабочих дней.
На подготовку итогового отчета по результатам ее проведения исполнителю отводится 30 рабочих дней, а на его направление заявителю — 3 рабочих дня. Копия такого отчета также направляется в Минпромторг. На основании отчета формируется окончательное заключение, которое в случае положительного характера сопровождается выдачей сертификата производителю лекарственного препарата.
Документы для сертификации
Чтобы получить сертификат GMP в России, производитель обращается в уполномоченный орган с заявлением, к которому прилагает пакет документов, включающий:
- копию документа, подтверждающего наличие у заявителя полномочий по взаимодействию с контролирующей организацией;
- копия основного досье используемого производственного объекта;
- информация о фактах несоответствия препарата действующим требованиям к качеству и безопасности и о фактах отзыва медикамента из оборота за период не менее 2 лет;
- полный список лекарств, который изготавливаются на данном производственном объекте;
- копия лицензии на производство лекарств;
- письмо о согласии на проведение инспекции производства.
Важнейшие документы предоставляются заявителем в копиях, поскольку при утере их восстановить невозможно или очень сложно. Правила регламентируют, что если заявление подает иностранный производитель, и некоторые документы в составе пакета представлены на другом языке, они должны быть переведены на русский язык и заверены в установленном порядке.
Сроки сертификации
Общая продолжительность процедуры сертификации складывается из следующих сроков.
|
Этап сертификационной процедуры |
Максимальная допустимая продолжительность |
|
Проверка полноты пакета документации, представленной с заявлением о сертификации, и правильности ее оформления, назначение инспекции |
10 рабочих дней |
|
Направление информации о назначении инспектирования в уполномоченное учреждение, которое проводит проверку |
3 рабочих дня |
|
Инспектирование и анализ лекарственного средства |
160 рабочих дней |
|
Принятие решения о выдаче заключения по результатам инспекционного отчета |
10 рабочих дней |
160-дневный период инспектирования включает внесение производителя в график инспекций, ожидание процедуры и проведение самой инспекции. Она должна занимать не более 10 рабочих дней.
Такой порядок действует, если в документации, поданной производителем, не обнаружат ошибок и недочетов, из-за которых ее могут направить на доработку. В этом случае вся процедура займет немногим более 180 рабочих дней, то есть свыше 8 месяцев.
Стоимость получения сертификата
Обязательной для всех производителей лекарственных средств, претендующих на получение сертификата, подтверждающего соответствие их продукции стандартам GMP, является оплата государственной пошлины за рассмотрение соответствующего заявления в Министерстве промышленности и торговли. Ее размер составляет 7500 рублей. Оплатить данную сумму необходимо еще до подачи заявления в ведомство, а ее размер никак не зависит от результатов рассмотрения документа.
Однако данная пошлина — это далеко не единственный и не самый крупный платеж, который потребуется осуществить производителю лекарств. Другой значительной статьей расходов станет плата за проведение экспертной оценки производства и продукции заявителя. Такая процедура выполняется специалистами ФГБУ «ГИЛС и НП»: для каждого из них предварительно проводится аттестация эксперта по GMP в России.
При этом размер платы за проведение оценки не является строго установленным, а определяется в зависимости от объема, характера и сложности необходимых процедур в соответствии с положениями приказа Министерства промышленности и торговли Российской Федерации от 11.01.2016 № 9 «Об утверждении методики определения размера платы за оказание услуги по инспектированию GMP». В случае, если проверка потребует проведения значительного объема работы и привлечения большого количества высококвалифицированных экспертов, размер платы за ее проведение может превышать 2,5 миллиона рублей.

Содержание
- Распространение стандарта GMP в фармацевтической сфере
- Международные, национальные и региональные правила GMP
- Появление стандарта GMP в России
- Стандарт GMP сегодня: в мире и в России
GMP — международный стандарт, определяющий нормы и правила, при соблюдении которых удается обеспечить высокое качество производственного процесса на всех этапах, в том числе связанных с хранением и испытанием продукции. Стандарт GMP включает ряд показателей, которым в обязательном порядке должно соответствовать предприятие, занимающееся выпуском той или иной продукции.
Ключевое отличие стандарта GMP от процедуры контроля качества заключается в том, что он имеет целостный подход, определяет параметры производства и лабораторной поверки «в целом». При этом процедура контроля качества направлена на исследование и оценку именно выборочных образцов (или их партий), определение их пригодности.
Распространение стандарта GMP в фармацевтической сфере
GMP — стандарт, который находит широкое применение в фармацевтической сфере, при производстве техники медицинского назначения. Также GMP совместно со стандартами GLP, GCP стандартизирует ряд параметров оценки качества медицинского обслуживания населения.
Начало активной борьбы за качество лекарственных средств пришлось на 1906 год, когда в США был принят Закон о доброкачественности пищевых продуктов и медицинских препаратов. С течением времени становилось очевидным, что проблемы качества лекарственных средств актуальны не только в Соединенных Штатах, но и далеко за их пределами.
Во многом это обусловлено стремительным развитием фармацевтического рынка во второй половине XX века. Именно в это время фармацевтический рынок впервые за историю стал обретать «глобальный характер», что вызвало необходимость в создании международных стандартов, дающих возможность:
- Унифицировать лекарственные средства.
- Регламентировать процесс производства ЛС.
- Установить нормы и правила хранения и распространения лекарственных препаратов.

Международные, национальные и региональные правила GMP
Создание и реализацию международного стандарта GMP по понятным причинам инициировали и поддерживали производители, которые боролись за честную конкуренцию, стремились к цивилизованному рынку, а также эффективному вложению капитала.
Так американская система GMP, регулирующая процесс производства лекарственных средств в одном государстве, получила колоссальную поддержку со стороны международных экспертов. И в 1968 году был разработан и утвержден первый официальный документ по уже международному стандарту GMP, над созданием которого работали специалисты Всемирной организации здравоохранения.
Спустя всего лишь год ВОЗ была принята резолюция, в соответствии с которой всем странам было рекомендовано применять стандарт GMP. В 1969 году к международной сертификации GMP присоединились 8 государств, а в 2019 году — уже более 40 стран используют национальные правила данного стандарта.
Впрочем, сегодня сертификация GMP имеет более «разветвленный характер», чем прежде. Например, выделяют несколько «градаций»:
- Международный стандарт GMP.
- Европейские правила GMP.
- Региональные правила GMP и т.д.
«Суммарно» более 140 стран на сегодняшний день участвуют в системе удостоверения качества лекарственных средств в международной торговле, которая базируется на соблюдении правил GMP.
Появление стандарта GMP в России
История появления системы GMP на территории Российской Федерации заслуживает отдельного внимания. В 1969 году властями нашей страны не была поддержана инициатива Всемирной организации здравоохранения. Вместо этого было принято решение о разработке собственного документа, который стандартизирует оборот лекарственных средств в соответствии с международными правилами. На его разработку ушло 5 лет: и в 1974 году в СССР были приняты рекомендательные правила производства лекарственных средств — РТМ 64-7-81-74. Спустя 7 лет они были пересмотрены, а процесс перехода к GMP стал неактуальным.
В 1991 году ЕС были утверждены новые правила стандарта GMP (они получили название GMP EU), которые распространялись на страны, входящие в Европейский союз. Одновременно с этим и власти Советского Союза приняли решение «уравнять» действующее на тот момент законодательство с международной практикой. Но реализовать такие планы удалось лишь после распада СНГ — только тогда «по примеру» международного опыта и началось создание собственной нормативной базы.
Российский GMP (фактически — аналог GMP EU) начал разрабатываться в 1998 году. Нашим правительством был утвержден план по поэтапному внедрению требований международного стандарта GMP на российских предприятиях, работающих в сфере фармацевтики. При этом срок реализации такого проекта был обозначен вполне конкретно — 2006 год. Но уже в 2004-ом был утвержден ГОСТ 52.249-2004 «Правила производства и контроля качества лекарственных средств».
Таким образом, нормы и правила международного стандарта GMP нашли свое «выражение» в собственном государственном стандарте ГОСТ Р 52249-2009, который вступил в силу 20 мая 2009 года. Данный документ распространяется на все категории лекарственных средств, определяет общие требования к их производству и оценке качества. Помимо этого, он устанавливает конкретные требования по изготовлению активных фармацевтических субстанций и некоторых видов лекарственных средств.

Стандарт GMP сегодня: в мире и в России
Важно отметить, что проверка на соответствие GMP лекарственных средств осуществляется не в добровольном, а в обязательном порядке. То есть обозначенные в стандарте правила и нормы должны неукоснительно соблюдаться фармацевтическими предприятиями.
На территории Российской Федерации за проверку на соответствие стандарту GMP отвечает «Государственный институт лекарственных средств и надлежащих практик» (это Федеральное бюджетное учреждение).
Отметим, в 2014 году был принял ФЗ №61 «Об обращении лекарственных средств». В соответствии с этим законом, всем национальным предприятиям, деятельность которых связана с производством медикаментов, в обязательном порядке было необходимо перейти на стандарт GMP, то есть обеспечить условия, при которых процесс изготовления будет полностью отвечать установленным правилам. Но на практике удалось реализовать это далеко не всем компаниям.
В настоящий момент только несколько десятков организаций смогли успешно обеспечить условия, при которых соблюдается GMP — производство лекарственных средств полностью соответствуют нормам и требованиям государственного стандарта качества.
Возвращаясь к международной практике, стоит сказать о том, что распространение системы сертификации GMP идет достаточно активными темпами. Например, весной 2017-го года пять государств Евразийского экономического союза объединились, начав работу в формате общего пространства, регулируемого едиными правилами надлежащей производственной практики ЕАЭС. В их число вошли: Россия, Беларусь, Армения, Казахстан и Кыргызстан.
Специалисты Gluvex знают все последние изменения в законодательстве, безупречно разбираются в лабораторном, аналитическом и технологическом оборудовании. Получите бесплатные консультации у специалиста по телефону: +7 (499) 270-16-62.
Лекарственные средства — продукция особой важности, поэтому к их безопасности требуется особый подход. Препараты низкого качества представляют собой не только опасность для здоровья, но и напрасную трату денег как для системы здравоохранения, так и для отдельных пациентов. Безопасность, эффективность и качество лекарственных препаратов обеспечивают стандарты GMP.
Что такое GMP
Надлежащая производственная практика (GMP — сокр. от англ. good manufacturing practice) — это система, которая обспечивает постоянный контроль фармацевтической продукции в соответствии со стандартами качества. Она предназначен для того, чтобы свести к минимуму риски, связанные с любым фармацевтическим производством, которые невозможно устранить, тестируя конечный продукт: неожиданное загрязнение готовых лекарств, неправильные этикетки на упаковках, недостаточное или слишком большое количество активного вещества, которые могут привести к неэффективному лечению или побочным эффектам.
GMP охватывает все аспекты производства: от исходных материалов, помещений и оборудования до обучения и личной гигиены персонала. Для каждого процесса, который может повлиять на качество лекарств, должны существовать прописанные инструкции и системы, обеспечивающие документальное подтверждение того, что правильные процедуры последовательно выполняются на каждом этапе производственного процесса — каждый раз, когда производится продукт.
Почему GMP — это важно
Как правило, покупатели не могут самостоятельно определить, безопасны ли лекарства и произведены ли они с надлежащим качеством. По умолчанию мы доверяем производителям в том, что лекарства соответствуют своим заявленным свойствам и не содержат вредных примесей. Лучшим подтверждением того, что компания выпускает качественные лекарства служит получение ей сертификата GMP.
Введение надлежащей производственной практики выгодно и самим производителям. Изготовление некачественной продукции приводит к значительным затратам. Вдобавок, в долгосрочной перспективе найти ошибки после того, как они были совершены, обходится дороже, чем предотвратить их изначально. Надлежащая производственная практика предназначена для предотвращения ошибок.
Кроме этого, речь идет о репутации производителей. Некачественные лекарства не только вредят пациентам, но и создают негативный имидж фармацевтических компаний. Производителям приходится отзывать серии проблемных лекарств с рынка, выделять команды для анализа ситуаций и потенциальных возмещений пациентам. Следование стандартам GMP позволяет значительно снизить риск таких ситуаций.
Основные требования GMP
- Производство и распространение лекарств должны минимизировать любой риск для их качества;
- Производственные помещения должны содержаться в чистоте, включая лаборатории и складские помещения;
- Производственные помещения, принципы работы и ее условия должны контролироваться, чтобы предотвратить загрязнение лекарственных препаратов;
- Все процессы должны быть четко определены и контролироваться для обеспечения согласованности. Любые изменения в производстве оцениваются с точки зрения безопасности пациента и качества продукта, а любые утвержденные изменения, которые могут повлиять на качество лекарственного средства, при необходимости аттестуются;
- Инструкции должны быть написаны четким и недвусмысленным языком;
- Во время производства и контроля качества должны быть сделаны записи, демонстрирующие, что все необходимые шаги были выполнены в соответствии с определением и что указанные характеристики качества продукции были соблюдены;
- Любые отклонения исследуются и документируются;
- Записи о производстве хранятся в понятном и доступном формате, позволяющем проследить всю историю партии лекарств;
- Есть рабочая система для отзыва любой партии из продажи или поставки;
- Жалобы на продаваемые продукты должны быть рассмотрены, причины дефектов качества расследованы, и должны быть приняты соответствующие меры в отношении дефектных лекарств и для предотвращения повторных нарушений.
Как регулируется GMP в мире
Стандарты GMP применяются к фармацевтическим производителям во всем мире, при этом они существуют сразу на нескольких уровнях регулирования: национальном и наднациональном, например, АТЭС (Азиатско-Тихоокеанском экономическом сотрудничестве), и других. У каждой страны, которая занимается производством лекарств, есть свои требования к фармацевтическим компаниям, но обычно они весьма похожи.
Большинство стран входит в Международный совет по гармонизации технических требований к лекарственным средствам для человека, основная задача которого состоит в выработке общих правил в производстве лекарств и унификации регуляторных актов. Поэтому нельзя сказать, что одни лекарства, если мы говорим о развитых странах, качественнее других. Практически все препараты в мире создаются и распространяются в идентичных условиях.
Как регулируется GMP в России
В России с 2014 года производство лекарственных средств на законодательном уровне обязано соответствовать стандартам GMP. Российский стандарт GMP был утвержден Приказом Министерства промышленности и торговли РФ от 14 июня 2013 г. № 916 “Об утверждении правил надлежащей производственной практики”. На сайте Минпромторга также можно проверить, какие производители имеют сертификаты GMP.
Поскольку GMP — обязательные правила, а не добровольные инструкции, то они подлежат проверке государством. В России проводить инспектирование иностранных производителей лекарственных средств для медицинского применения на соответствие стандартам уполномочено Федеральное бюджетное учреждение “Государственный институт лекарственных средств и надлежащих практик”.
При этом соответствовать российским стандартам должны компании, которые не имеют производства внутри страны, но поставляют в нее лекарства. Иными словами, отечественные эксперты проверяют любую площадку, включая иностранную, лекарства с которой доходят до полок аптек в России. Поэтому у большинства производителей есть сразу несколько сертификатов GMP от стран, где они продают лекарства.
Технически, производить лекарства в России без сертификата возможно, но только в двух случаях. В первом варианте речь идет о производстве без реализации, то есть готовые препараты никогда не дойдут до покупателя. Второй вариант предполагает выпуск лекарств на экспорт, что запрещает продавать их в России. В таком случае компании нужно иметь сертификат GMP той страны, куда планируется вести поставки. Но, учитывая схожесть регулирования надлежащей производственной практики во всем мире, в этом случае стандарты производства все равно придется поддерживать на должном уровне.
Нужно не забывать, что большинство производителей в России имеют еще и сертификат GMP ЕАЭС. С 2022 года регистрация по правилам Евразийского экономического союза стала обязательной для производителей, которые находятся на его территории. Правда, для производителей это не составляет проблем, так как требования в новом регулировании идентичны требованиям, предъявляемым Минпромторгом России, но формулировки дополнительно гармонизированы с требованиями GMP ЕАЭС.
Как получить сертификат GMP в России
Любой производитель, который хочет подтвердить свое соответствие надлежащей производственной практике в России, должен пройти ряд процедур. Начинается все с подачи заявления в Минпромторг России, специалисты которого проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
После этого информация передается в “Государственный институт лекарственных средств и надлежащих практик”, который, в случае отсутствия проблем с документацией, определяет дату инспекции. Согласно законодательству, она должна пройти в течение 160 дней после подачи заявления. В целом, вся процедура занимает в среднем 8 месяцев, а проходить ее требуется раз в 3 года.
Текст: Владимир Пучнин
.
Post Views:
1 281
Повышение квалификации GMP
GMP — это список требований, которые предъявляются к предприятиям и организациям, занимающимся производством лекарственных средств и контролем их качества. Специалисты компаний должны знать эти правила, чтобы соответствовать действующим профессиональным стандартам. Для них в СНТА разработаны соответствующие курсы повышения квалификации. Обучение проводится в онлайн-режиме, благодаря чему слушателям не нужно лично присутствовать на лекциях и отрываться от основной работы.
Описание курса
Повышение квалификации по GMP предполагает самостоятельное изучение слушателями учебных планов, разработанных для конкретных направлений. В них рассматриваются такие вопросы, как:
- Организация фармацевтического производства;
- Обеспечение и контроль качества препаратов;
- Основные разделы GMP;
- Проведение аудита предприятия на соответствие GMP и многие другие.
Для тех, кто проходит обучение GMP, подготовлены комплекты электронных учебников. Они размещены в Личных кабинетах слушателей, доступ к которым предоставляется сразу после подтверждения зачисления.
Почему стоит выбрать СНТА для повышения квалификации?
- Удобный и доступный каждому онлайн-формат обучения;
- Помощь технической службы;
- Методическое обеспечение;
- Консультации преподавателей;
- Зачисление без экзаменов;
- Доступная стоимость.
Получаемые документы
Повышение квалификации по GMP заканчивается проверочным онлайн-тестом, по результатам которого высылается удостоверение установленного образца. Оно принимается в РФ.
Условия поступления
Для зачисления в Академию нужно подать заявление, подписать договор, внести оплату и выслать копии паспорта и диплома о высшем или среднем специальном образовании. Чтобы подать заявку, позвоните нам по номеру 8 (800) 707-48-27.